





Stigende strategisk betydning
Pharma 09/2023 - Serie: Fra molekyle til medicin
Virksomhedernes regulatory affairs-afdelinger (RA) sikrer godkendelse af lægemidler, hvilket indebærer dialog med myndighederne fra den tidlige udviklingsfase over godkendelsesprocessen og til videreudvikling og vedligehold af produktet. RA får stigende strategisk betydning, og der kommer løbende ny, skærpet lovgivning, som RA-afdelingerne skal forholde sig til.
Af Christian K. Thorsted
Lægemiddelvirksomheders mål er konstant at udvikle nye behandlinger, der hjælper patienterne, og som giver økonomisk grundlag for fortsatte forskningsinvesteringer.
Det kræver, at nye produkter får en markedsføringstilladelse, som skal vedligeholdes i hele deres levetid – opgaver som løses af virksomhedernes regulatory affairs-afdelinger.
Tidligere var det en funktion, som kunne anses som lidt kedelig, hvor gigantiske papirdynger til sidst i forløbet blev indsamlet og indsendt til lægemiddelmyndighederne.

Men i moderne life science-virksomheder er funktionen alt andet end kedelig og hengemt, og papiret er naturligvis fortid.
”Dokumentation og guidelines kan lyde tørt, og måske har der også tidligere været en oplevelse af, at regulatory affairs – qua deres rolle som gatekeeper mellem virksomheden og myndighederne – var besværlige at have med at gøre," siger farmaceut Hanne Arentsen, karrierevejleder i Pharmadanmark, og fortsætter:
"RA skal oversætte myndighedernes krav til en godkendelse til en strategi, som virksomheden skal køre efter i et projekt. Det kan i nogle tilfælde også betyde, at RA må påpege, at der er noget i et projekt, myndighederne aldrig vil gå med til – det kan måske være upopulært, men nødvendigt."
Hun har over 20 års erfaring fra forskellige RA-job i lægemiddelindustrien, blandt andet i Novo Nordisk, LEO Pharma og senest som Senior Regulatory Strategy Leader i Lundbeck.
Strategisk spiller
I den tid har hun bemærket flere markante forandringer i virksomhedernes RA-funktion.
”I starten af mit arbejdsliv som regulatorisk medarbejder synes jeg, vi skulle kæmpe lidt mere for at få strategisk indflydelse. Vi blev lidt opfattet som dem, man kunne konsultere, når der skulle skrives dokumenter til ansøgningerne. Med tiden synes jeg dog, at RA er blevet inviteret ind tidligere og tidligere, og nu i høj grad er en vigtig strategisk partner gennem hele lægemiddeludviklingen," fortæller hun.
Hun har selv i et tidligere job besøgt et lille biotekfirma og set på dets tidlige in vitro og prækliniske data, altså før firmaet overhovedet havde startet de humane kliniske forsøg.
"Allerede på det tidspunkt gav det nemlig rigtig god mening at overveje de regulatoriske aspekter, hvis vi skulle gå ind og finansiere firmaets projekter. Der var brug for en vurdering af, om firmaet havde lavet de studier, som opfylder myndighedernes krav til den prækliniske pakke. Jo tidligere RA kommer ind, jo hurtigere kan man komme med en vurdering af, om den regulatoriske vej frem er nem eller svær.”
Hanne Arentsen mener, at det interessante i RA er, at det er en funktion, hvor man kan være med i den strategiske udvikling hele vejen igennem.
"Der er naturligvis også nogle vigtige funktioner indeholdt i RA, som er mere operationelle end strategiske, men jeg tror, at mange bliver inden for RA, fordi man har en følelse af at sidde midt i beslutningsprocessen, hele vejen, og der hvor tingene sker.”
Life science-branchen er reguleret af et utal af regler og myndighedskrav, der påvirker mange funktioner i en virksomhed. Derfor er regulatory affairs altid en del af en større sammenhæng, og et RA-team samarbejder på tværs af fagligheder.
Det betyder, at der mere end nogensinde er behov for medarbejdere med helhedsforståelse af aktiviteter og processer forbundet med at få lægemidler på markedet, påpeger Hanne Arentsen.
Stigende krav
Hun bemærker også, at kravene til dokumentation i forbindelse med lægemiddelgodkendelse kun er øget fra myndighedernes side.
Som eksempel er der stigende fokus på generering af Real World Evidence (RWE) til at supplere data fra de randomiserede, kontrollerede kliniske forsøg – altså data der indsamles fra befolknings- eller patientgrupper, og som blandt andet kan give mulighed for at evaluere, hvordan patienter reagerer på bestemte behandlinger i praksis og uden for rammerne af et kontrolleret klinisk forsøg.
”RWE kan bidrage til forståelsen af anvendelsen af en medicinsk behandling i almindelig klinisk praksis, og det har en afsmittende effekt på udviklingen af lægemidler. For pludselig er det ikke længere kun vigtigt at kende til effekten i randomiserede kliniske forsøg, altså under meget styrede betingelser i meget homogene patientpopulationer. Det bliver tiltagende vigtigere at kende til effektiviteten ude i den virkelige verden. Hele diskussionen om, hvordan man belyser begge aspekter er meget interessant og diskuteres intensivt i disse år," peger Hanne Arentsen på.
Men det forholder sig ikke sådan, at brug af RWE så betyder, at myndighederne slækker på nogle af de andre krav, for eksempel til randomiserede kliniske forsøg.
"Så der er blevet lagt mere på. Der er hele tiden nye krav, som giver mening, men som øger kompleksiteten for en RA-afdeling i forhold til den dokumentation, som er nødvendig for markedsføringstilladelse.”
Der er da også sket en voldsom eksplosion i antal medarbejdere inden for RA.
Da det regulatoriske arbejde begyndte at tage form i slutningen af 1970’erne, var det muligt for alle regulatoriske medarbejdere i danske virksomheder at sidde rundt om ét mødebord i den daværende industriforening MEFA (Foreningen af danske Medicinfabrikker).
Pharmadanmarks lønstatistik for 2023 viser, at RA sidenhen er blevet et meget stort ansættelsesområde.
15 procent af de medlemmer, der har svaret på lønstatistikken, angiver, at de arbejder inden for RA (214 medlemmer) – kun overgået af medlemmer inden for kvalitetssikring/QA (22 procent) og Forskning & Udvikling (21 procent).
Ligeledes er der kommet en videnskabelig tilgang til det regulatoriske område, såkaldt Regulatory Science; her beskæftiger man sig med, hvordan lovgivningen kan påvirkes og implementeres, ligesom der i disciplinen ligger en forståelse for det regulatoriske områdes voksende betydning for medicinbrugerne og virksomhederne.
En overbevisende pakke
Det ultimative mål for RA er at sikre en regulatorisk godkendelse ved at kunne indsende en overbevisende og fyldestgørende dokumentationspakke, som kan godkendes af myndighederne.
”Myndighederne vurderer på sikkerhed, effekt og kvalitet, når de skal godkende produkter. Det vil sige, at de selvfølgelig ser på, om de prækliniske- og kliniske studier er lavet korrekt, ligesom de vil have sikkerhed for, at de sikkerhedssignaler, der kommer ud af studierne, bliver reflekteret rigtigt i dokumentationen – og at produktet er udviklet og fremstillet i henhold til lovgivningen," siger Hanne Arentsen.
Med hensyn til effekten går myndigheden meget op i, om produktet faktisk repræsenterer en forbedring i forhold til det, der allerede er på markedet. Så den del er man i RA også meget opmærksom på, at der er tilstrækkelig dokumentation for. Der skal påvises et klinisk merværdi.
RA hjælper til med at tilpasse dokumentationen og er undervejs i dialog med myndighederne. Man kan også få inspiration til en strategi ved konstant at følge med i, hvad konkurrenterne gør.
Hvis en konkurrent for eksempel har opnået markedsføringstilladelse med et fase 3 studie med 300 patienter, kan man måske selv prøve en lignende strategi.
Så det handler også om hele tiden at følge med i, hvad myndighederne godkender, og hvad de afviser. På den måde bliver RA også en meget central spiller i for eksempel designet af kliniske studier.
”En guideline er jo en retningslinje, og man vil altid over for myndighederne forsøge at komme igennem med det, der passer bedst til virksomhedens strategi; og trykteste myndighederne en lille smule på de krav, de stiller,” siger hun.
Helt fast struktur
Hanne Arentsen fortæller, at regulatory affairs arbejder inden for en helt fast struktur; Common Technical Document (CTD) (se figur 1).
Denne trekant er international standard for indsendelse af dokumentation til regulatoriske myndigheder stort set verden over. Det betyder, at man allerede tidligt i udviklingsprocessen kan begynde at bygge sin dokumentationspakke op.
”RA har en rigtig vigtig funktion, for det er for sent at opdage mangler i dokumentationspakken tæt på indsendelsen. Derfor arbejder RA fra dag 1 strategisk med opbygning af dokumentationen efter de guidelines, som findes på de pågældende områder. Hvis man ikke formår at lave en overbevisende dokumentationspakke, så risikerer man, at lægemiddelkandidaten ikke kommer på markedet eller forsinkes,” understreger Hanne Arentsen.
Det betyder, at det undervejs i udviklingsprocessen er vigtigt at have så meget dialog med myndighederne som muligt, så man har en klar fornemmelse af deres forventninger til udviklingen og dokumentationen.
”Man bruger lang tid på at planlægge denne strategiske dialog, og forudse, hvad de vil svare. Ofte har man en god idé om, hvor dokumentationspakken har sine svagheder, men der kommer altid noget uforudset og nødvendig viden ud af at konsultere myndighederne.”
Ligeledes bruger man meget tid på at prøve at forudse alle de spørgsmål, man kan få fra myndighederne, når man indsender sin dokumentation og på at lave svarstrategier til disse spørgsmål.
Langt hovedparten af det, man forestiller sig at kunne blive spurgt ind til, kommer aldrig nogensinde i spil, er Hanne Arentsens erfaring.
”I RA bliver man ofte spurgt ’hvad tror du myndighederne vil sige’? Der skal man så lige kigge i sin krystalkugle. Det er det, der gør regulatorisk strategi rigtig svært, men også fagligt spændende og udfordrende,” slutter hun. 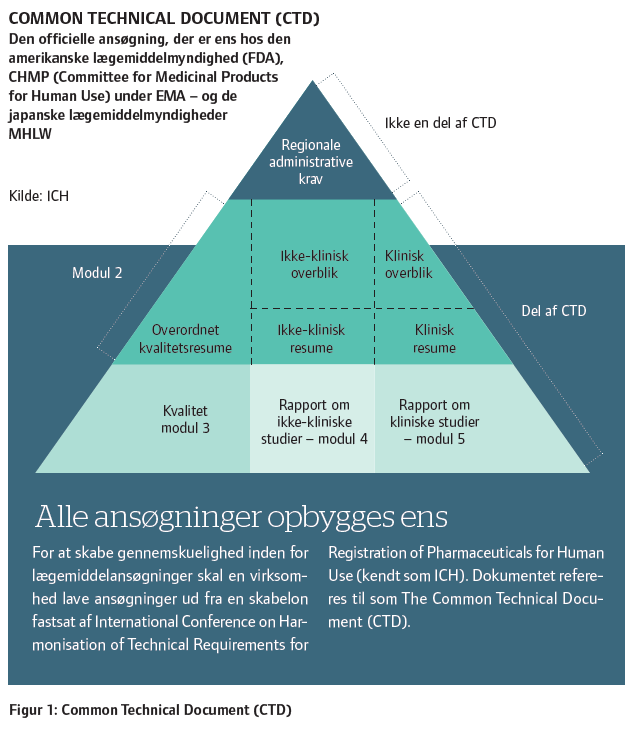
| Novo Nordisk Fonden har uddelt en fem-årig bevilling på op til 50 millioner kroner til International Collaborative Bioscience Innovation & Law (Inter-CeBIL) Programme ved Københavns Universitet. CeBil så dagens lys i 2018 og kan nu fortsætte i et udvidet format under navnet Inter-CeBIL. Baggrunden for etableringen af CeBIL er, at det bliver stadigt mere komplekst at bringe nye lægemidler og biosolutions fra laboratoriet til markedet. Det er således afhængigt af stadig mere komplekse love og regulatoriske processer. Derfor har CeBIL skullet levere indsigt, viden og værktøjer, der bidrager til et vellykket, konkurrencedygtigt og bæredygtigt innovationsøkosystem. ”Med Inter-CeBIL ønsker vi nu at bygge videre på det, vi har lært, og samtidig skifte fokus fra den indflydelse, vi har haft på et højt niveau, til mere anvendelige og håndgribelige effekter på et mere basalt niveau,” siger forskningsleder professor Timo Minssen fra Centre for Advanced Studies in Biomedical Innovation Law (CeBIL) på Det Juridiske Fakultet ved Københavns Universitet. Med den nye finansiering sigter Inter-CeBIL i mod at styrke og understøtte et konkurrencedygtigt biovidenskabeligt innovationsøkosystem ved at undersøge juridiske og regulatoriske udfordringer. Inter-CeBIL vil på baggrund af bevillingen nu udvide sit forskningssigte og sine samarbejder og byde flere nøglepartnere fra Danmarks Tekniske Universitet (DTU) velkommen sammen med flere forskningsstipendiater og personer tilknyttet topuniversiteter fra hele verden. |
Læs de andre artikler i kapitlet Fra molekyle til medicin – regulatory affairs og pharmacovigilance:



